The SPInDel is an alternative approach for biological identification based on the length of ribosomal RNA (rRNA) gene regions. In general, alignments of primary rRNA gene sequences from different species show alternating regions of nucleotide conservation and variation, both in terms of nucleotide substitutions (commonly called “SNPs”) and insertion/deletion (indel) events. The presence of indels results in sequences of different lengths and introduces gaps in the alignment, typically denoted by a dash “-”.
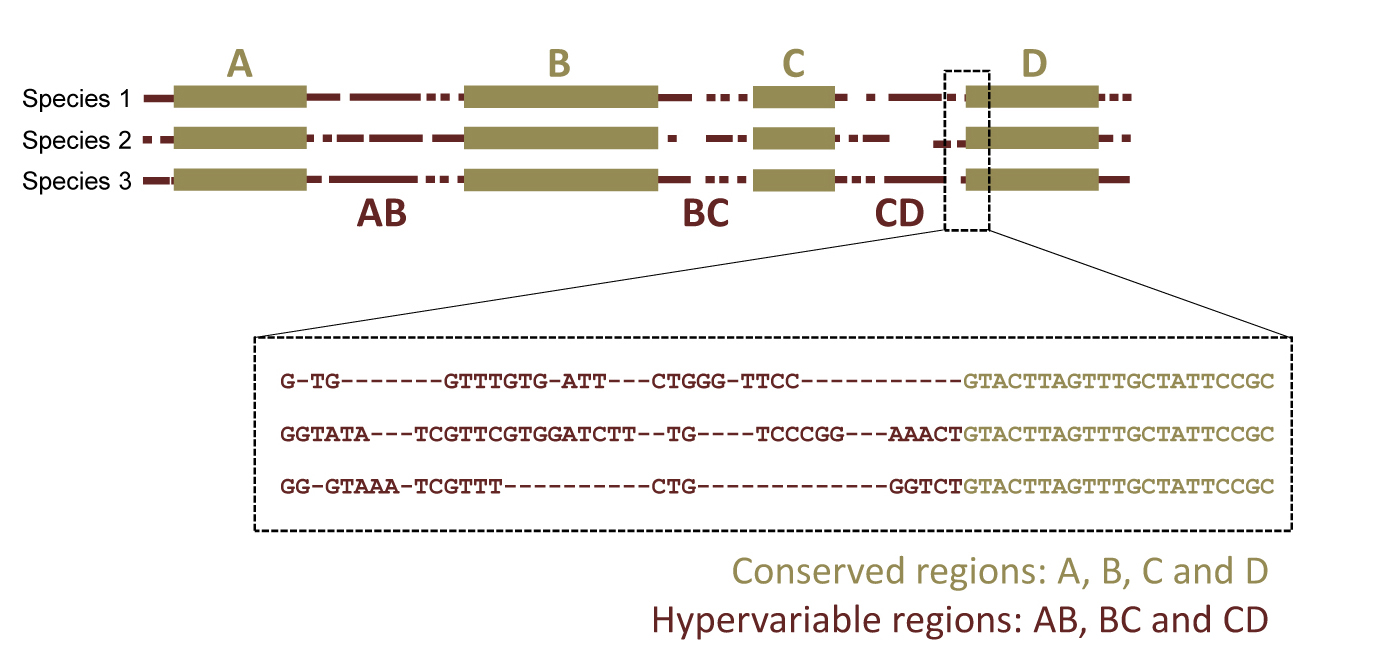
Our concept for biological identification uses rRNA gene sequences as follows: conserved regions are used to define variable segments (“SPInDel hypervariable regions”) in which a combination of sequence lengths is characteristic of each species (a “SPInDel profile”). Thus, each species can be defined by a unique numeric profile.
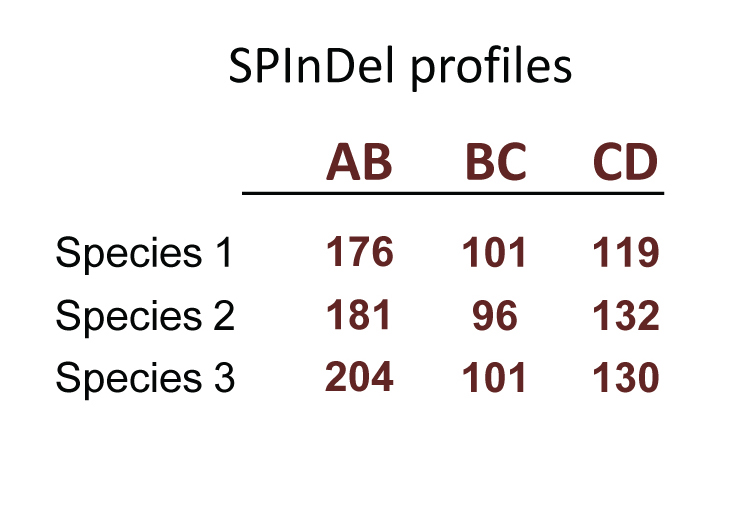
In theory, a survey of just 6 hypervariable regions with 20 alleles each (or 11 regions with 5 alleles each) is enough to discriminate all eukaryotic species on Earth, which are estimated to be between 5 and 15 million in number. In practice, SPInDel is able to discriminate 93.3% of eukaryotic species with low intraspecific variation and high phylogenetic resolution (for general statistics on current projects click here).
Other genomic regions may present similar patterns of sequence evolution and thus may also be suitable for species identification using the SPInDel concept (for instance, in viruses). Detailed information can be found in this publication.